Theory of Drug Design
What are drugs?
The vast majority of drugs are small molecules designed to bind, interact, and modulate the activity of specific biological receptors. Receptors are proteins that bind and interact with other molecules to perform the numerous functions required for the maintenance of life. They include an immense array of cell-surface receptors (hormone receptors, cell-signaling receptors, neurotransmitter receptors, etc.), enzymes, and other functional proteins. Due to genetic abnormalities, physiologic stressors, or some combination thereof, the function of specific receptors and enzymes may become altered to the point that our well-being is diminished. These alterations may manifest as minor physical symptoms, as in the case of a runny nose due to allergies, or as life threatening and debilitating events, such as sepsis or depression. The role of drugs is to correct the functioning of these receptors to remedy the resulting medical condition.
As an example, the highest grossing drug in 2000 was Prilosec, which earned $4.102 billion in sales. Prilosec is used to treat stomach ulcers and acid reflux disease. Prilosec targets a specific enzyme, the proton pump, which is located in the acid producing cells lining the stomach wall. This enzyme is responsible for the production of stomach acid. Due to genetic reasons, such as deficient enzymes that regulate acid secretion, or physiologic causes, such as stress, too much acid may be produced. This leads to ulceration of the stomach lining or acid reflux disease and heartburn. Prilosec binds to the proton pump and shuts it down, thereby diminishing the production of stomach acid and its associated symptoms.
The biochemistry of drugs
Enzymes are a subset of receptor-like proteins that are
directly responsible for catalyzing the biochemical reactions that sustain
life. For example, digestive enzymes act to break down the nutrients of
our diet. DNA polymerase and related enzymes are crucial for cell division
and replication. Enzymes are genetically programmed to be absolutely
specific for their appropriate molecular targets. Any errors could have
grave consequences. One can imagine the end result should blood clotting
enzymes start activating throughout the body. Or consider the problems
that arise when our immune system attacks our own tissues. Enzymes ensure
the specificity of their targets by forming a molecular environment that
excludes interactions with inappropriate molecules. The analogy most often
mentioned is that of a lock and key. The enzyme is a molecular lock, which
contains a keyhole that exhibits a very specific and consistent size and
shape. This molecular keyhole is termed the active site of the
enzyme and allows interaction with only the appropriate molecular targets.
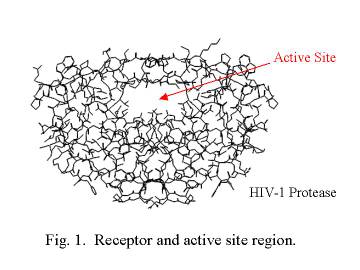
Just as a typical lock is much bigger than the keyhole, the receptor is usually
much larger than the active site. The receptor, as specified by our DNA,
is a folded protein whose major purpose is to form and maintain the size and
shape of the active site. This is illustrated in Figure 1 using the
structure of the HIV-1 protease.
The most important concept in drug design is to understand the methods by which the active site of a receptor selectively restricts the binding of inappropriate structures. Any potential molecule that can bind to a receptor is called a ligand. In order for a ligand to bind, it must contain a specific combination of atoms that presents the correct size, shape, and charge composition in order to bind and interact with the receptor. In essence, the ligand must possess the molecular key that binds the receptor lock. Figure 2 schematically shows a typical ligand-receptor binding interaction.
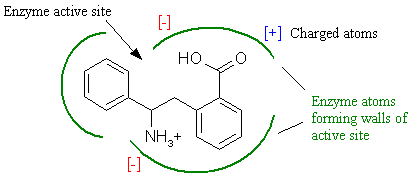
Figure 2. Enzyme substrate complementary interactions.
We see here that a putative ligand-receptor interaction must have complementary size and shape. This is termed steric complementarity. As is the case with an actual key, if a different molecule varies by even a single atom in the wrong place, it may not fit properly, and will most likely not interact with the receptor. However, the more closely the fit between the ligand and receptor, the more tightly the interaction becomes. Again, keep in mind that this is only a two-dimensional schematic. Both ligand and active site are volumes with complex three-dimensional shape.
In addition to steric complementarity, electrostatic interactions influence ligand binding. Charged receptor atoms often surround the active site, imparting a localized charge is specific regions of the active site. From physics, we know that opposite charges attract while similar charges repel. Electrostatic complementarity further restricts the binding of inappropriate molecules since the ligand must contain correctly placed complementary charged atoms for interaction to occur.
The main driving force for ligand and receptor binding is hydrophobic interaction. Nearly two-thirds of the body is water, and this aqueous milieu surrounds all our cells. In order for ligand and receptor to interact, there must be a driving force that compels the ligand to leave the water and bind to the receptor. The hydrophobicity of a ligand is what causes this. Hydrophobicity stands for 'water fearing' and is a measure of how 'greasy' a compound is. It can be roughly approximated by the percentage of hydrogen and carbon in the molecule. This force is easily demonstrated by placing a few drops of oil in a cup of water. The oil is composed of hydrocarbon chains and is highly hydrophobic. The oil droplets will instantly coalesce into a single globule in order to avoid the water, which is highly polar. As shown in Figure 2, the active site may contain a mixture of hydrophobic pockets and regions that are more polar. Since the hydrophobic portions of the ligand and receptor prefer to be juxtaposed, the arrangement of hydrophobic surfaces provides yet another way that receptors can limit the binding of inappropriate targets.

As discussed above, there are numerous potential interactions between ligand and receptor. Depending upon the size of the active site, there may be a myriad steric, electrostatic, and hydrophobic contacts. However, some are more important than others. The specific interactions that are crucial for ligand recognition and binding by the receptor are termed the pharmacophore. Usually, these are the interactions that directly factor into the structural integrity of a receptor or are involved in the mechanism of its action. Using our lock and key analogy from above, we can imagine a lock having numerous tumblers. There may be many keys that can sterically complement the lock and fit within the keyhole. However, all but the correct key will displace the wrong tumblers, leading to a sub-optimal interaction with the lock. Only the correct key, which presents the pharmacophore to the receptor, contacts the appropriate tumblers and properly interacts with the lock to open it. This is crucial to the design of pharmaceuticals since any successful drug must incorporate the appropriate chemical structures and present the pharmacophore to the receptor.
This is shown in Figure 3 above. In the upper left frame of this figure, we see our native ligand bound within the active site. Assume that through biochemical investigation, we determine that the phenyl ring (blue) and the carboxylic acid group (green) are vital to receptor interaction. Thus, we deduce that these two groups must be the pharmacophore that a ligand must present to the receptor for binding. In future drugs that we develop to mimic the native ligand, we must include these two pharmacophoric elements for successful binding to occur. This is shown in the upper right derivative compound where a bicyclic group has been substituted. Because it maintains the pharmacophore and retains its complementary size and shape, it has a reasonable chance of successfully binding. However, any drug that we develop which lacks a complete pharmacophore may not interact with the receptor target.
The challenge of drug design
Given our introduction to the biochemistry of ligand receptor binding, we can begin to appreciate the difficulties in designing drugs towards specific target receptors. Table 1 lists the major tasks and concerns in this endeavor.
|
Table 1. Major tasks and concerns in drug development. |
|
1. Characterize medical condition and determine receptor targets. |
|
2. Achieve active site complementarity: steric, electrostatic, and hydrophobic. |
|
3. Consider biochemical mechanism of receptor. |
|
4. Adhere to laws of chemistry. |
|
5. Synthetic feasibility. |
|
6. Biological considerations. |
|
7. Patent considerations. |
When a medical condition exists where a drug could be beneficial, extensive scientific study must first be done in order to determine the biological and biochemical problems that underlie the disease process. This often takes years of study in order to characterize the targets for a potential drug. The reason is that nearly all biological processes in the human body are tightly interconnected. Altering the behavior of select receptors or enzymes may have detrimental effects with other systems. These are the side effects that occur with nearly all drugs. Furthermore, the human body is a homeostatic machine, and always attempts to achieve equilibrium. As a result, the body will attempt to counteract any pharmacotherapeutic intervention.
Once a receptor target has been established
and well characterized, the process of ligand design begins. Obviously,
the first consideration is that the designed ligand must complement the active
site of the receptor target. Steric, electrostatic, and hydrophobic
complementarity must be established as we discussed above. The
pharmacophore must be presented to the receptor in order for recognition and
binding to occur. Otherwise, the designed ligand will have no chance of
interacting with the receptor.
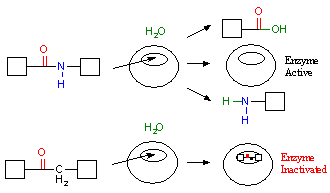
Figure 4. Designing ligands to offset enzyme mechanism.
In addition to adequately binding the receptor, the biochemical mechanism of the receptor target must be taken into consideration. This is shown in Figure 4. In this figure we schematically represent the biochemical mechanism of a protease. A protease is an enzyme that cleaves proteins and peptides. In the top part of the figure, we see that a protease recognizes a specific group of atoms, colored in red and blue, called a peptide bond. If the peptide bond is present at a specific position in the active site when the ligand binds, it is cleaved by the protease with the addition of water (H2O) to form two separate fragments. If our goal is to inactivate this protease, any designed ligand cannot possess this peptide bond at the same position. Otherwise, it will simply be cleaved by the protease, and the protease will continue to function unperturbed. However, the ligand can be modified so that the peptide bond is no longer present as shown in the bottom portion of the figure. If this ligand is then bound by the enzyme, the enzyme will not be able to cleave it. As such, the enzyme would be inactivated, as the ligand remains lodged in the active site.
Having characterized the active site region and the mechanism of action of the target receptor, the challenge then becomes one of designing a suitable ligand. This is, by far, the most daunting task of the entire drug design process. The optimal combination of atoms and functional groups to complement the receptor is often the natural ligand of the receptor. Unfortunately, this is usually an unacceptable candidate for a drug. This is because the natural ligand either fails to inactivate the receptor, as described above, or it is a natural substance that cannot be patented. Patent considerations are often paramount, as legal protection for the developed drug affords the opportunity to recoup the financial costs of development. Therefore, alternate combinations of chemical structures must be devised.
The design of novel ligands is often restricted by what chemists are physically able to synthesize. It is of no use to design the ultimate drug if it cannot be manufactured. The laws of chemistry dictate that each atom type has a specific size, charge, and geometry with respect to the number and types of neighboring atoms that it can be joined to. The entire field of chemistry is predicated on the establishment of synthetic rules for the construction and manipulation of various combinations of atoms and functional groups. It is the expertise in these chemical rules that govern the ability of the synthetic chemist to design and synthesize postulated ligand candidates. Within these rules, the drug developer must creatively propose suitable chemical structures that satisfy the requirements discussed above.
Finally, there are biological considerations to the development of new drugs. The liver is the major organ of detoxification in the human body. Any drug that is taken undergoes a number of chemical reactions in the liver as the body attempts to neutralize foreign substances. This set of reactions is well characterized, and a great deal of knowledge exists as to how drugs are modified as the body eliminates them. More importantly, various chemical structures are highly toxic to biological systems, and these are also well characterized. These constraints must also be taken under consideration as novel drugs are developed.
The drug discovery pipeline
As discussed above, the development of any potential drug begins with years of scientific study to determine the biochemistry behind a medical problem for which pharmaceutical intervention is possible. The result is the determination of specific receptor targets that must be modulated to alter their activity in some way. Once these targets have been identified, the goal is then to find compounds that will interact with the receptors in some fashion. At this initial stage of drug development, it does not matter what effect the compounds have on the targets. We simply wish to find anything that binds to the receptor in any fashion.
The modern day drug discovery pipeline is outlined in Figure 5. The first step is to determine an assay for the receptor. An assay is a chemical or biological test that turns positive when a suitable binding agent interacts with the receptor. Usually, this test is some form of colorimetric assay, in which an indicator turns a specific color when complementary ligands are present. This assay is then used in mass screening, which is a technique whereby hundreds of thousands of compounds can be tested in a matter of days to weeks. A pharmaceutical company will first screen their entire corporate database of known compounds. The reason is that if a successful match is found, the database compound is usually very well characterized. Furthermore, synthetic methods will be known for this compound, and patent protection is often present. This enables the company to rapidly prototype a candidate ligand whose chemistry is well known and within the intellectual property of the company.
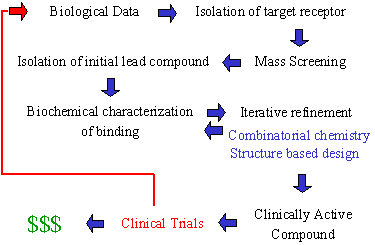
Fig. 5. Drug discovery pipeline (circa 2001).
If a successful match is found, the initial hit is called a lead compound. The lead compound is usually a weakly binding ligand with minimal receptor activity. The binding of this structure to the receptor is then studied to determine the interactions that foster the ligand-receptor association. If the receptor is water soluble, there is a chance that x-ray crystallographic analysis can be employed to determine the three-dimensional structure of the ligand bound to the receptor at the atomic level. This is a very powerful tool for it allows scientists to directly visualize a snapshot of the individual atoms of the ligand as they reside within the receptor. This snapshot is referred to as a crystal structure of the ligand-receptor complex. Unfortunately, not all complexes can be analyzed in this manner. However, if a crystal structure can be determined, a strategy can then be developed based upon this characterization to improve and optimize the binding of the lead compound. From this point onward, a cycle of iterative chemical refinement and testing continues until a drug is developed that undergoes clinical trials. The techniques most often used to refine drugs are combinatorial chemistry and structure based design.
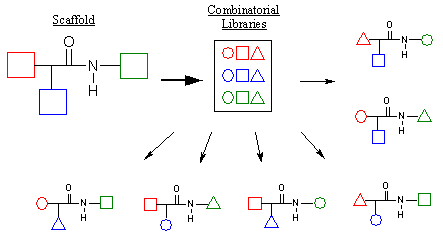
Figure 6. Combinatorial chemistry schematic.
Combinatorial chemistry is a very powerful technique that chemists can employ to aid in the refinement of the lead compound. Combinatorial chemistry is a synthetic tool that enables chemists to rapidly generate thousands of lead compound derivatives for testing. As shown above in Figure 6, a scaffold is employed that contains a portion of the ligand that remains constant. Subsite groups (shown in red, green, and blue) are potential sites for derivatization. These subsites are then reacted with combinatorial libraries to generate a multitude of derivative structures, each with different substituent groups. One can see how a vast number of compounds can be generated as a result of the combinatorial process. If a scaffold contains three derivatization sites and the library contains ten groups per site, theoretically 1000 different combinations are possible. By carefully selecting libraries based upon the study of the active site, we can target the derivatization process towards optimizing ligand receptor interaction.
![]()
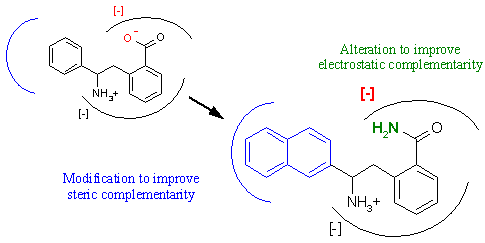
Structure based design, often called rational drug design, is much more focused than combinatorial chemistry. As shown above in Figure 7, it involves using the biochemical laws of ligand-receptor association discussed above to postulate ligand refinements to improve binding. For example, we discussed that steric complementarity is vital to tight receptor binding. Using the crystal structure of the complex, we can target regions of the ligand that fit poorly within the active site and postulate chemical changes to improve complementarity with the receptor. In a similar fashion, functional groups on the ligand can be changed in order to augment electrostatic complementarity with the receptor. However, the danger in altering any portion of the ligand is the effect on the remaining ligand structures. Modifying even a single atom in the middle of the ligand can drastically change the shape of the overall structure. Even though complementarity in one portion of the ligand might be improved by the chemical revision, the overall binding might be severely compromised. This is the difficulty in any ligand refinement procedure.
Use of computers in drug design
In the early 1990s there was a great deal of optimism that computer aided drug design would revolutionize the way in which drugs could be developed. The enduring exponential increase in computing power had progressed to the point that rudimentary estimations of ligand receptor complementarity could be performed. Furthermore, computer graphics technology had achieved the ability to generate vector models of chemical structures and manipulate them in real-time. This offered, for the first time, the ability to interactively study computer models of ligand structures and their binding interactions with a receptor.
Concomitant with the development of this technology was the emergence of the AIDS epidemic. During the late 1980s, scientists had isolated the causative agent of AIDS, the HIV-1 virus. Considerable characterization of the viral life cycle provided numerous potential targets for pharmaceutical intervention. Among them was the HIV-1 protease. This aspartyl protease was an enzyme that was unique to HIV, and absolutely required for the processing and maturation of HIV proteins. Thus, if a drug could be developed to inactivate this protease, the virus would be unable to generate mature infectious particles to sustain the infection. Numerous groups around the world rapidly solved the crystal structure of this enzyme (see Figure 1). The mechanism of this enzyme was determined, and the layout of the active site was carefully mapped.
It was known that humans possess similar classes of proteases. Renin is an enzyme secreted by the kidneys that is responsible for initiating a cascade of reactions that regulate blood pressure. It too is an aspartic protease, and ligands that inhibited its function were known. With the wealth of data from the study of the HIV-1 protease, the hope was that this target could be exploited by computer-aided rational drug design to rapidly generate novel AIDS drugs. Computational chemists believed they could circumvent much of the time and effort required for drug synthesis and testing by simply generating novel compounds using the computer. Testing would be replaced by merely calculating the ligand-receptor binding affinity using the physical laws of chemistry. The concept of generating virtual lead compounds entirely through computer simulation was termed denovo design.
Difficulties implementing denovo design
Many of the worlds largest pharmaceutical firms spent millions of dollars on hardware and software in their endeavor to make denovo design a reality. Unfortunately, successes were rare. Except for a few exceptions, denovo design was an utter failure, and did not prove to be an effective method to discover lead compounds. The main reasons were limitations in computing power and the lack of useful software functionality. In scientific computing, accuracy and processing time are always a tradeoff. Thus, in order to make the calculations run in a finite period of time, a plethora of assumptions, significant approximations, and numerous algorithmic shortcuts had to be utilized. This, in turn, greatly diminished the calculated accuracy of any ligand receptor interaction. As such, chemists could postulate numerous chemical structures that could potentially complement the active site; however, the calculated binding had no correlation with reality.
This remains the most significant challenge in denovo design to this day. Although computers have become exponentially faster, the sheer number of calculations needed to accurately predict the binding of a denovo generated ligand to its receptor in a useful timeframe still requires significant approximations. In denovo design, we are attempting to generate a whole ligand from scratch and dock it within the receptor. As stated above, the difficulty lies in predicting how the chemical structure will behave in real life. A ligand is an inherently flexible structure, and can assume a plethora of different conformations and orientations. The big question remains whether the predicted binding structure will mirror the calculated one. Failure in this endeavor has undermined the utility of denovo structure generating software. We will discuss these shortcomings and the technological advances of RACHEL, which attempt to circumvent these deficiencies, in detail below.
The second most significant problem in computer aided denovo design is the generation of undesired chemical structures. There are a nearly infinite number of potential combinations of atoms. However, the vast majority of these structures are of no use. As discussed above, undesired structures are rejected due to toxicity, chemical instability, or synthetic difficulty. Nearly all denovo design software packages are plagued by this problem, especially with respect to synthetic feasibility. Thus, although such software can postulate potential complementary ligands, the vast majority of them are worthless. We will discuss in great detail below how RACHEL attempts to circumvent this problem with newly developed technology.
The end result of these shortcomings was that computer aided denovo design soon fell out of favor as a means of generating viable lead compounds. By the mid 1990s there had been a tremendous number of denovo software packages released; however, they all suffered these same problems. Gradually, such programs were shelved and investigators looked to other technologies to aid in their drug development efforts.
Rebirth of computer-aided drug refinement
It was at this time that the techniques of mass screening and combinatorial chemistry began to gain widespread acceptance and use. The use of mass screening and combinatorial chemistry allowed researchers to discover lead compounds in a rapid and efficient manner. As such, denovo design tools and their associated problems were no longer needed to generate lead structures. One would surmise that computer-aided drug design technology would have soon ceased to exist. On the contrary, it soon became apparent that computational tools were needed that could optimize these lead compounds into potent drugs.
The concept of drug optimization versus denovo design is an important one. The difficulty with denovo ligand generation is that an entire structure is being created from scratch. The confidence one has of accurately predicting how this structure will interact and bind within a target receptor is shaky at best. In drug optimization, we begin with a lead compound whose bound structure within the receptor has been characterized, most likely through x-ray crystallography. Subtle modifications are then performed to generate derivative compounds using structure based drug design to improve binding affinity. Because we are making much smaller changes, our faith in the validity of the resulting structures is far greater. These derivatives then undergo testing to determine which modifications improve binding. The structures of the best ligands can then be elucidated to verify the accuracy of the modifications. This refinement process continues iteratively until optimal binding ligands are produced.
Since subtle modifications are being made to a common structure, the predictive ability of ligand refinement software is much higher. This is because the effect of a single chemical modification on ligand-receptor binding is far easier to quantitate than an extreme change. No longer are we trying to determine the binding affinities of drastically different structures. Instead, we are simply determining the rank order of a list of derivative compounds. This greatly increases the confidence that proposed structures will bind in a manner consistent with our understanding.
In addition, the act of generating chemical derivatives is highly amenable to computerized automation. Consider the application of targeted structure based combinatorial chemistry as discussed above. Libraries of derivative components are assembled based upon the analysis of the active site. Because of the combinatorial nature of this method, an extremely large number of candidate structures may be possible. A computer can rapidly generate and predict the binding of all potential derivatives, creating a list of the best potential candidates. In essence, the computer filters all weak-binding compounds, allowing the chemist to focus, synthesize, and test only the most promising ligands. Thus, utilizing computer aided drug design software to aid in the refinement of weak binding lead compounds is the most effective manner in which these tools can be employed. The use of computer modeling to refine structures has become standard practice in modern drug design.
Rational drug design software - State of the art
Since the 1990s, dozens of rational drug design packages have been published. These programs fall in one of three main genres: 1. Scanners, 2. Builders, or 3. Hybrids.
Scanners

Figure 8A. Overview of database search programs.
All database search programs fall into this category. Scanner type programs are more or less used for lead compound screening. Figure 8A shows how these programs are utilized. In the upper left corner, we see an active site with a lead compound whose binding structure has been determined. From biochemical analysis of the ligand-receptor interaction, we determine that three ligand groups make up the pharmacophore: a phenyl ring (green), an amide hydrogen (blue), and a hydroxyl group (red). The pharmacophore is transformed into a query that specifies the three-dimensional relationship between the various functional groups desired. A database of compounds is then utilized whose three-dimensional structures are known. The query is then used to search the database for compounds that mimic the pharmacophore and can potentially bind to the receptor target. Example results are shown in Figure 8B below.

Figure 8B. Database search results.
Database search programs have inherent strengths. To begin, the user has complete control over the query specifications. This allows for the retrieval of structures that meet the requirements of the pharmacophore and have a better opportunity to complement the receptor. Secondly, because these programs utilize a database of known compounds, synthetic feasibility is not an issue. In addition, these programs are usually highly optimized for speed, which allows for the rapid determination of potential binding ligands. Finally, since compounds are retrieved that mirror the query, no scoring functions are required. The assumption is that the three-dimensional structure stored in the database is representative of biological reality. Although this can be true of small molecules, larger structures are often too flexible for the assumption to hold true.
Database search programs also have inherent weaknesses that limit their application. First and foremost, a database of structures is required. Although pharmaceutical companies are likely to maintain corporate databases, many academic institutions lack the manpower and funding to generate this wealth of data. Secondly, the database has a finite number of structures, which limits and biases potential solutions. Finally, there is no recombination or derivatization of retrieved structures, which builder-type programs exhibit. Thus, the diversity of potential hits is limited as well.
Builders
Although builder-type programs may be used for denovo ligand design, their chief utility is in the optimization of lead compounds. These programs also utilize a database of structures; however, the database contains fragments and chemical building blocks instead of complete compounds. In order to use a builder-type program, the user must possess a lead compound that requires optimization. Such compounds normally contain an area that poorly complements the corresponding receptor region. This is illustrated in the upper left corner of Figure 9. Here we see a lead compound that contains a stable, tight-binding region (black), and a phenyl ring that should be replaced to improve receptor complementarity (red). Builder-type programs require the attachment point of the weak-binding portion as input. The software then removes the offending ligand region and uses the attachment point to create a population of derivatives by adding, deleting, and substituting fragments chosen from the component database (green) to fill the active site. The binding energies of the resulting derivative ligands are then calculated. Those structures that augment binding are retained while those that do not are discarded. This process repeats as the new population of structures is then processed to generate the next round of derivatives. By making incremental changes iteratively, these programs generate a set of ligands with improved receptor complementarity over time.
Figure 9. Demonstration of
Builder-type programs.
There are several advantages in using these types of programs. First, no database of structures is required, allowing anyone to use these programs without additional requirements. Although a component database is utilized, this is often built into the software. The combinatorial addition of fragments offers a vast number of potential derivative structures. Because components from numerous chemical classes are included, these programs automatically generate a diverse set of chemical solutions. This also contributes to the creation of truly novel ligands. Finally, these programs lend themselves well to the current drug development pipeline, since they can be utilized to optimize the hits that result from mass screening.
However, the strengths described above for builder-type programs lend to their weaknesses. With the combinatorial attachment of such diverse chemical components, the generation of synthetically unfeasible structures is commonplace. Furthermore, ligands that are chemically unstable are produced as well. Although a diverse set of chemical building blocks are utilized, the manner in which they are attached is entirely up to the developer of the software. Decisions such as when a particular component is selected and where it is attached greatly affect the generated structures. These choices reflect the bias of the program developers. In addition, the success of any builder program to generate improved ligands is entirely dependent upon its ability to accurately calculate the receptor-binding structure and binding affinity. As we have stated above, this is a very challenging problem.
Conformational searching in drug design
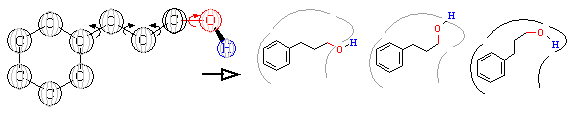
Figure 10A. Ligand flexibility.
Ligand flexibility is a concern in
builder-type programs. The ligand analogy to a key discussed above is slightly
misleading. While a key is a static unbending object, a chemical structure
is constantly changing shape. As shown in Figure 10A, a molecule is
actually composed of rigid chemical groups separated by rotatable bonds, as
defined by the laws of chemistry. These rotatable bonds give ligands
inherent flexibility. In the right side of this figure, we see how a
ligand can adopt numerous configurations as it attempts to bind within the
active site. Given a newly generated compound, the big challenge is to
computationally search all the potential configurations to determine if binding
is possible. If so, the task is then to determine the most complementary
binding structure.
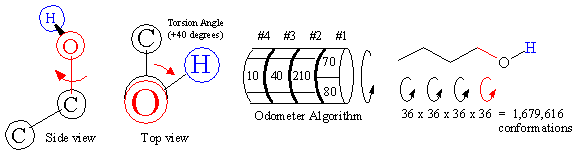
Figure 10B. Combinatorial nature of conformational searching.
Although accurately calculating ligand receptor affinity remains the most scientifically challenging endeavor in drug design, searching all the rotatable bond configurations a ligand can adopt is the most demanding. A snapshot structure of the ligand at any instant in time is called a conformation, and is defined by the set of torsion angles between rigid groups. As shown in Figure 10B above, each rotatable bond can potentially spin 360 degrees, and the two attached groups define the torsion angle. The difficulty in determining all possible conformations lies in the combinatorial nature of the problem. Because each bond can potentially sweep the entire arc, an 'odometer' algorithm must be employed.
The odometer algorithm is a systematic
sampling of all possible torsion angle combinations. Like an odometer, the
first bond is fully rotated 360 degrees before the second bond is
incremented. When the second bond is incremented, the first bond is reset
and then fully scanned again. This continues until the second bond is fully
rotated, at which time the third bond is incremented. Searching continues
in this manner until all rotatable bond combinations are eventually sampled as
shown above.
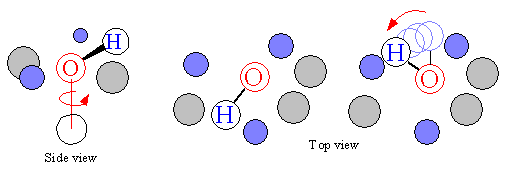
Figure 10C. Determining valid conformer torsion angles.
During the conformational search, acceptable torsion angle ranges must be elucidated for each rotatable bond. This is shown in Figure 10C. When a rotatable bond is incremented, the atoms attached to the 'swing arm' are checked against all receptor atoms and ligand groups within the vicinity. If contact exists, then that particular conformer is eliminated. As such, only valid ligand conformations that conform to the active site are generated.
As shown in Figure 10B above, the combinatorial nature of this problem leads to an exponential rise in the number of conformations that must be calculated. A four rotatable bond ligand sampled at ten-degree increments adds up to 1,679,616 different conformations. However, a five rotatable bond ligand sampled at ten-degree increments includes over 60 million conformations! Considering that most drugs contain 10 - 15 rotatable bonds, this value can easily overwhelm even the fastest computers.
Algorithms have been developed that reduce the computational burden of conformational searching by orders of magnitude. Nevertheless, commercial builder packages implement conformational flexibility in different ways to compensate for the associated computational burden. Some do not implement conformational flexibility at all, which severely limits their ability to determine adequate ligand binding conformations. Others use rudimentary, pre-calculated torsion angle scans that lack the resolution to tightly dock compounds within the active site. Only packages that implement a systematic conformational search are able to sufficiently determine potential binding conformations.
Hybrids
The primary utility of a hybrid program is
in denovo ligand generation. Figure 11 depicts how these programs
work. In the upper left hand corner of this figure, we see an active site
with three distinct regions colored in blue, green, and red. The goal of a hybrid program is to generate a
complete ligand that complements the active site region. To do so, these
programs employ a combination of scanner and builder algorithms in divide and
conquer approach. These programs first utilize scanner technology to find
components that will complement individual subsites within the active site
volume. This is depicted by the components in the blue, green, and red boxes. These individual components are then
docked into their respective regions within the active site as shown in the
upper right. Splicing fragments, shown in purple, are then used to join these components into a
single complete ligand. It is important to note that numerous possible
fragments may exist that complement the various active site regions. Thus,
a potentially large number of ligands may be generated by combinatorially
linking the various components.
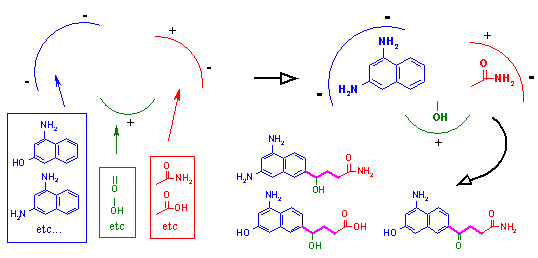
Figure 11. Overview of hybrid-type programs.
As stated above, hybrid-type programs are mainly used for denovo generation of lead compounds. The strength of these programs is in their ability to generate a large number of diverse potential hits. However, they suffer the same shortcomings as all denovo design packages described above. The difficulties in accurately calculating ligand receptor binding affinity are significant. In addition, the combinatorial nature of the algorithm often leads to the generation of chemical structures that are undesirable, unstable, or synthetically difficult. Finally, the developer of the software may bias the generation of compounds. For example, many of these programs place components within the active site using a pre-determined binding algorithm based upon the functional groups that are presented by the receptor. Additionally, the algorithms used to splice the components together greatly affect the generated ligands.
RACHEL software package
RACHEL stands for Real-time Automated Combinatorial Heuristic Enhancement of Lead compounds and is a drug optimization package written by Chris M.W. Ho, M.D.Ph.D. of Drug Design Methodologies, LLC. This program is designed to optimize weak binding lead compounds in an automated, combinatorial fashion. From our discussions above, this software would be classified as a 'builder-type' drug refinement program. With our brief introduction into the basics of drug design, we can now discuss the technological advances that make RACHEL superior to all current computer-aided drug refinement software packages.
Extraction of building blocks from corporate databases
We have discussed how builder-type programs work. In short, a database of chemical fragments is used to derivatize a lead compound by replacing weak binding regions with components that will improve receptor complementarity. These compounds are then scored by calculating their affinity for the receptor. Those compounds that bind tightly with the receptor are then saved while those that bind poorly are discarded. This new population of compounds is then processed to form the next generation of derivatives. Over time, a lead compound is iteratively refined into a set of tight binding structures.
When using current commercial ligand refinement packages of this type, the user is dependent on the software for several vital functions that are critical for drug development success. First and foremost, a database of chemical building components is required. All current commercial packages provide this database of components, which allows any researcher to immediately use these tools for drug design. However, this is not always desirable. Pharmaceutical firms are always competing against rival companies. Intellectual property in the form of patented database structures, synthetic know-how, and the biochemical data of characterized lead compounds provides a competitive edge against other companies. As such, using a program that contains both a standard database and a standard scoring function offers no such advantage over another company.
RACHEL avoids this shortcoming by allowing the company (or academic laboratory) to utilize its intellectual property in the design of new drugs. RACHEL pulls building block components directly from the users corporate database. As such, companies who have invested considerable time and money developing the chemistry to synthesize a particular class of compounds can utilize this knowledge in the design of future drugs. This has the added benefit that generated structures are easier to patent as proprietary chemistry is incorporated. This is a tremendous advantage to using our software, and no other design package employs this feature.
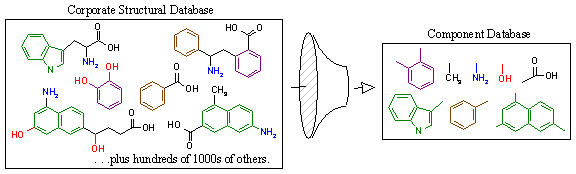
Figure 12A. Extraction of components from corporate structural database.
Figure 12A schematically demonstrates this process. On the left, we have the corporate structural database, which may contain hundreds of thousands of compounds. All structures are composed of non-rotatable chemical groups separated by rotatable bonds as defined by the laws of chemistry. These non-rotatable groups represent the fundamental building blocks that will be used to regenerate new derivative compounds. RACHEL first isolates these components by identifying the rotatable bonds in the structure as shown below in Figure 12B (red arrows). The individual components are then isolated, identified with a unique label that describes its distinct chemical architecture, and stored in the component database along with a description of its chemical composition. The component label is very important as it is used to register each fragment and prevent the storage of redundant chemical groups.
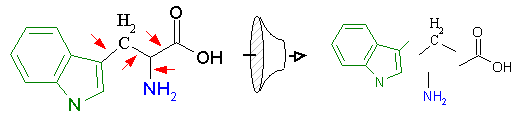
Figure 12B. Separation and isolation of components at rotatable bonds.
There are numerous advantages to extracting components in the manner described above. First, the storage of unique components allows the compression of a massive corporate database into a much smaller and manageable form. Typically a corporate database containing 100,000 structures may be comprised of only 5,000 individual components. This is because a few select components, such as methyl, hydroxyl, and amine groups, are utilized over and over again. Second, unique chemical constructions, for which only proprietary synthetic methods are known, are stored and available for use in future ligand design. This allows the user to take advantage of patented corporate chemistry and preserve the competitive edge gained from prior research.
Intelligent Component Selection System
The goal of builder-type programs is to generate derivatives that are complementary to the active site. Both steric (size and shape) as well as electrostatic forces must be considered. The difficulty in accomplishing this lies in the sheer number of potential component combinations that are possible. As a result, nearly all commercial packages randomly select fragments for assembly. While this ensures an adequate sampling of components, it often leads to the selection of improper fragments. As such, many iterations of structure refinement are wasted generating poor derivatives.
The RACHEL software has a far greater problem. While current builder-type software packages contain databases with 100 components or less, RACHEL can extract upwards of 40-50,000 components depending upon the size and diversity of the corporate database. Thus, the number of potential fragment combinations is nearly immeasurable. Clearly, a method is needed to rapidly focus on the appropriate combinations that are likely to satisfy binding requirements.
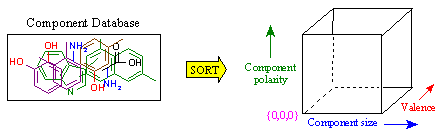
Figure 13A. Generation of Diversity Index.
The greatest benefit of RACHEL's component extraction method is that a massive diversity index of the entire corporate database is created. Along with the atomic coordinates of each component, a wealth of chemical information characterizing each building block is stored. Data such as the size of the component, atom composition, connectivity, ring structure, and electrostatic charge are included. As such, a means of rapidly cross-referencing chemical components on demand is available.
Figure 13A demonstrates how this diversity index is generated. On the left, we see a representative component database. Using the stored chemical attributes, the database is sorted and mapped into a multi-dimensional array, where each axis represents a different descriptor. In this example, we only show size, polarity, and valence (number of connections) for simplicity. Each axis provides a gradient along which components can be distinguished. As such, components that are similar with respect to the various descriptors are grouped together.
This diversity index offers a powerful means to improve the generation of complementary ligands. Over time, builder-type programs evolve compounds with improved binding. A moderate affinity structure has reasonable steric and electrostatic complementarity with the active site. However, components can still be added, deleted, or substituted to augment receptor interaction.
As stated above, nearly all commercial builder-type programs select substituent fragments at random. Although simplistic, this is absolutely necessary to ensure adequate sampling of the database and generate truly novel solutions. RACHEL implements random sampling in the initial stages of lead compound optimization. Early derivatives that are generated are weak binding at best. Thus, random component sampling increases the chances of finding the appropriate components to improve receptor interaction.
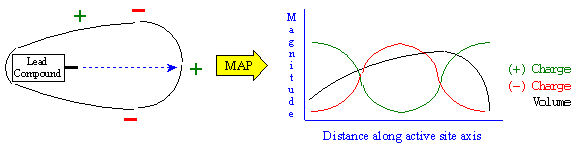
Figure 13B. RACHEL Active Site Mapping.
However, random sampling often diminishes the complementarity of reasonable binding compounds. This is the result of replacing satisfactory components with poor ones. For example, if a small methyl group or a highly charged fragment were to replace a large, hydrophobic ring on the ligand, it would ruin interaction with the receptor at that component. Instead, RACHEL incorporates a heuristic active site mapping algorithm as shown in Figure 13B to determine the optimal chemical characteristics to complement a given region of the active site. This technique maps chemical characteristics of the receptor, such as positive charge, negative charge, and active site volume as a function of distance along the active site axis. Using this active site map, RACHEL can determine the chemical characteristics most likely to complement the receptor at a given component location. RACHEL then determines a list of candidate fragments and substitutes them in a combinatorial fashion.
This is diagrammed in Figure 13C below. In this example, RACHEL determines that the naphthalene group (blue) and carboxylic acid group (red) of a ligand derivative should be replaced with other components to improve binding. The naphthalene group is large and very non-polar since it is strictly hydrocarbon. Conversely, the carboxylic acid group is quite small, but highly polar. Using the active site map as described above, RACHEL determines that these characteristics are indeed ideal for complementing the receptor at each respective component. Using the diversity index, RACHEL can cross-reference other database components that exhibit similar characteristics, as shown in the red and blue boxes on the right. These components are then combinatorially used to generate a new family of derivatives for testing. Each derivative retains the optimal receptor binding characteristics. However, enough variability is generated to potentially improve receptor complementarity.
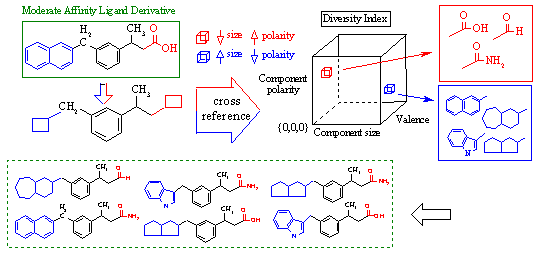
Figure 13C. RACHEL Intelligent Component Selection System.
Development of a component specification language
User-directed structure generation
The ability to instantly cross-reference components by chemical composition also permits user-directed structure generation, which is the most powerful and unique feature of RACHEL. No other builder-type drug design package incorporates this feature. Essentially, this technology permits the true application of virtual combinatorial chemistry. The inspiration for this technology stems from earlier work published by the author - 'DBMAKER: A set of programs to generate three-dimensional databases based upon user-specified criteria' Ho, CMW., and Marshall, GR., J. Comp-Aided Mol. Design, 9:65-86 (1995).
Figure 14A demonstrates this with an example. In the middle of this figure, we see a lead compound scaffold containing an amide bond with various side chains extending from it.
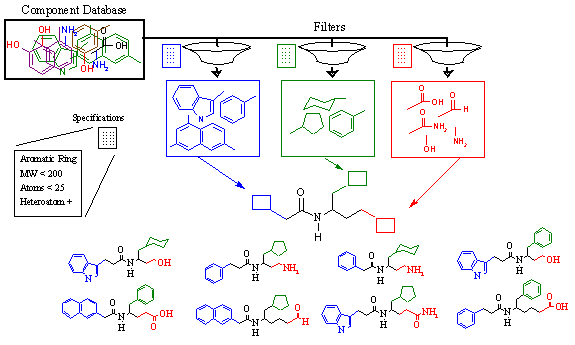
Figure 14A. RACHEL user-directed structure generation.
From biochemical characterization of this lead compound we discover that three chemical groups make up the pharmacophore. The first group, shown in blue, must contain a large ring system. Crystallographic analysis reveals that both single and bi-cyclic rings are capable of binding, as long as they are planar. Thus, they must be aromatic. Any atom types may be accepted. The second group, shown in green, has different requirements. Again, a cyclic component is desired. However, the binding pocket in this region is smaller, but more spherical. Thus, only single rings are acceptable although they need not be aromatic. In addition, this region is very hydrophobic; thus, only hydrocarbon components are acceptable (only carbon and hydrogen). The third group, shown in red, is quite different from the first two. This region of the active site is highly charged, and requires a small polar group to interact with. Thus, no ring structures are acceptable. Furthermore, heteroatoms (nitrogen, oxygen) are required.
|
BLUE derivatives |
GREEN Derivatives |
RED Derivatives |
|
(+) Ring structures - aromatic |
(+) Ring structure - single |
(-) Ring structures |
|
Molecular weight < 200 |
Molecular weight < 100 |
Molecular weight < 50 |
|
# Atoms < 25 |
# Atoms < 20 |
# Atoms < 8 |
|
(+) Any atom type |
(+) C, H = only |
(+) N, O = required |
Table 5. Chemical requirements for each
derivative group in Fig 14A.
The various chemical requirements of each derivative group are summarized in Table 5. In order to implement these requirements, a component specification language has been developed. This specification language contains a combination of keywords, target values, and Boolean operators. A brief summary of these commands is listed in Table 6 below. The specification language is very powerful and allows the user to control these characteristics and many more. Once the chemical requirements are established for each derivative group, RACHEL then filters the master component database and generates individual databases for each subsite.
|
Command |
Function |
|
CMPNTS min - max |
Number of total components to utilize. |
|
ATOMS min - max |
Number of atoms in a specific component. |
|
R-ATOMS min - max |
Number of ring atoms in a specific component. |
|
MW min - max |
Molecular weight. |
|
LINKS atypes (<,>,=) value |
Specifies rotatable bond atom types between components. |
|
ATYPES (list) (<,>,=) value |
Specifies atom type requirements in a specific component. |
|
BONDS (list) (<,>,=) value |
Specifies bonded atom types within a specific component. |
|
PHARM (atype) {x,y,z} |
Specifies a specific pharmacophoric group at coordinates {xyz}. |
Table 6. RACHEL component specification language.
Using these individual databases, shown in Figure 14A as the blue, green, and red boxes, RACHEL combinatorially generates all possible derivatives within the constraints of the active site. In so doing, an immense number of diverse chemical structures may be constructed and tested in a defined and controlled manner.
Filtration of components using constraints
As we have demonstrated, the user has considerable control over the chemical species that can be incorporated into structures. However, another feature unique to RACHEL that the component specification language permits is the removal of undesired structures. As stated above, this is one of the shortcomings in computer-aided drug design, especially with builder-type programs. Builders can often generate combinations of components that are neither stable nor synthetically feasible. The terms listed in Table 6 may be used in a variety of ways to limit the generation of these unacceptable structures. This is illustrated in Figure 14B below.
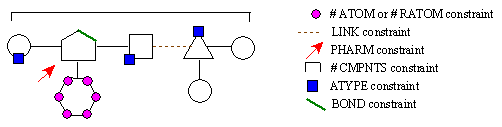
Figure 14B. RACHEL component constraints.
We see here the various constraints RACHEL can employ and how they relate to a given structure. In this hypothetical chemical structure, the geometric shapes represent different components while the lines connecting them correspond to rotatable bonds. The ATOM and RATOM constraints govern how many atoms a particular component can possess. The LINK constraint limits the atom types that can be utilized in rotatable bonds. The PHARM specification signifies that a specific atom type must be present at a precise location in the active site. The #CMPNTS restriction places upper and lower bounds on the total number of components a structure can possess. The ATYPE constraint stipulates how many atoms of a specific type can be present in both individual components as well as the entire structure. The BOND specification places limits on the bonded atom types that can be present within a component. As one can see, this again gives the user a tremendous amount of control over the structures generated by RACHEL.
Template driven structure generation
An additional feature that is unique to RACHEL is an automated method to generate diversity using templates. These templates are an integral part of the component specification language. As is normally the case, a computational chemist is not sure exactly what derivative components might complement the receptor. However, specific chemical groups may be desired at general locations in the active site. These might be pharmacophoric elements, or proprietary chemical structures for which synthetic methods are available. This is illustrated in Figure 14C below.
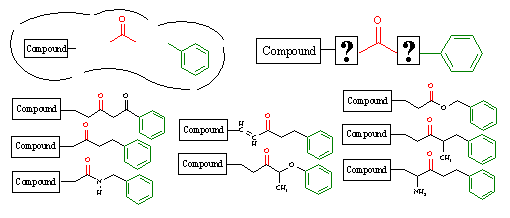
Figure 14C. Template driven structure generation.
In the upper left, we show an active site that contains a portion of a lead compound that has been previously characterized. We also know that two chemical groups, a carbonyl group (red) and a phenyl ring (green), are required to satisfy the pharmacophore for ligand binding. Given these issues, RACHEL allows the user to define a chemical template, as shown in the upper right of this figure, to generate appropriate structures. In this template the lead compound fragment and the two pharmacophoric groups are separated by wildcard designations, which denote where chemical variability can occur.
RACHEL will then generate chemically diverse structures using the template as shown in the figure above. The static portions of the template are left untouched, and they are incorporated into every generated derivative. However, the wildcard regions allow RACHEL to creatively insert various components in a random manner to link these pharmacophoric elements together. Obviously, constraints can be placed on these variable regions using the component specifications described above. As such, the use of these templates enables RACHEL to fully explore the chemical diversity within the corporate database and maintain the fundamental groups necessary to achieve receptor binding.
Novel techniques to estimate ligand - receptor binding
Automated Scoring Function Elucidation
As we discussed above, the calculation of receptor binding affinity for each newly generated derivative ligand remains the most challenging aspect of drug design. Not only is this task very difficult, it also is critical for the success of the program. If the calculated binding affinities have no bearing on reality, then, the program might as well generate random structures. The big difficulty is that the accurate determination of ligand receptor binding involves complex, intensive, quantum chemical calculations, which can take days to weeks on even the fastest computers. On the other hand, a typical ligand refinement program can generate and sample new structures at the rate of several hundred to thousands a minute. Thus, in order to achieve such high throughput, there must be some compromise in the accuracy of the binding calculation. That compromise is in the utilization of scoring functions.
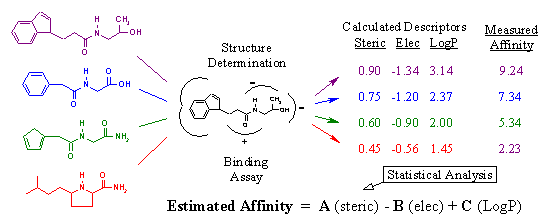
Figure 15A. Derivation of scoring function.
Scoring functions estimate ligand-binding affinity using descriptors that can be rapidly measured from the ligand receptor interaction. In essence, a scoring function is an equation that relates measurable descriptors of binding to ligand receptor affinity. Figure 15A reveals how scoring functions are derived. Given a particular ligand and receptor, two things must be done. First, the ligand must be reacted with the receptor to determine its actual binding affinity using a biological assay. Second, the three-dimensional structure of the ligand bound within the receptor must be determined using x-ray crystallography. As we discussed above, the determinants of binding include steric interaction energy, electrostatic interaction energy, and hydrophobicity. Given the three-dimensional structure of a particular compound bound within the active site, we can rapidly calculate the values for these descriptors. For review - see Head, R.D. et. al, J. Am. Chem. Soc., 118: 3959-3969 (1996).
For example, to calculate steric interaction energy, we simply sum up the number of receptor atoms that are within a specific distance (i.e. 5 angstroms) away from any ligand atom. The higher the value, the more interaction between ligand and receptor atoms. Electrostatic interaction energy is computed using Coulombs law, which can be found in any high school physic book. Hydrophobicity is represented by LogP, which is a measure of the compounds propensity to solubilize in oil versus water. The higher the value, the more greasy and oily the compound is. In short, these descriptors are simple and very easy to calculate. This allows for the rapid determination of characteristics that relate to ligand binding strength.
In our figure, we have four complexes whose binding affinity has been measured and whose descriptors have been calculated. Statistical tools, such as partial least squares regression, are then employed to generate the equation relating the numerical trends in the descriptors with the corresponding binding affinities. What results is an equation where estimated affinity is a function of the calculated descriptors (steric, electrostatic, and logP) multiplied by their corresponding coefficients (A, B, and C). It is the coefficients that relate the calculated descriptors to the actual affinities, and are determined by the statistical analysis. For example, we can see that as steric interaction energy increases, so does the biological binding activity. Thus, the coefficient (A) is positive. On the other hand, we know that a negative electrostatic interaction energy is conducive to tighter binding since opposite charges attract. Therefore, the corresponding coefficient (B) is negative. LogP follows a similar trend as steric interaction energy; thus, coefficient (C) is positive.
Once a scoring function has been derived, it can be employed to estimate binding affinities very rapidly. Given a newly designed ligand or structural derivative that has been docked within the active site, the descriptors of binding are first calculated. These descriptors are then multiplied by the derived coefficients of the scoring function. Once all the terms have been calculated, they are then summed to determine the estimated binding affinity of the ligand in question. It is important to note that this example is very simplistic. In reality, some scoring functions contain over twenty terms. Table 7 lists the ligand receptor binding descriptors employed by RACHEL.
|
Steric complementarity. |
Molecular weight. |
|
Steric strain. |
Number of rotatable bonds. |
|
Electrostatic interaction energy. |
LogP estimation. |
|
Nonpolar - nonpolar interaction energy. |
Nonpolar atom fraction. |
Table 7. RACHEL descriptors used to generate scoring functions.
Currently, there are several hundred high quality ligand-receptor complexes in the public domain that can be employed for scoring function development. Pharmaceutical firms have access to far more proprietary structures. However, even with all these structures along with the powerful statistical tools to analyze them, scoring functions still remain mediocre at best. According to the laws of thermodynamics, DG = DH - TDS. DG is the Gibbs free energy of binding, and is the energy that is released when ligand and receptor bind. This is the actual thermodynamic property that we are trying to estimate with the scoring function. DH is enthalpy (internal energy), and is grossly approximated by the calculated descriptors. Efforts to improve the accuracy of these approximations often increase calculation time drastically. TDS is an entropy term, and is indicative of the relative gain or loss of disorder when ligand and receptor bind. Perhaps the biggest influence on entropy is the behavior of the water molecules in the active site that are displaced when binding occurs. This is often disregarded, as it is difficult to accurately calculate without considerable computation. Thus, the take-home message is that DG is at best very crudely estimated by any scoring function. For review - see Ajay and Murcko, J. Med. Chem., 38: 4953-4967 (1995).
Compounding this problem is the fact that all current commercial packages utilize a single, proprietary, generalized scoring function that has been derived using a wide variety of structures. There are two significant problems with this approach. First, receptor systems vary considerably in their chemical makeup. In some systems, electrostatic interactions dominate the ligand binding force. In other systems, hydrophobic interactions overshadow the other forces involved. Thus, a master scoring function to estimate binding affinity for all ligand receptor systems becomes a 'jack of all trades, master of none'. Using such a variety of ligand receptor systems in the training set adds considerable noise to the data, which diminishes its predictive power.
The second shortcoming in using a generalized scoring function is the loss of competitive advantage. In effect, any laboratory or company that employs these tools utilizes the same predictive function as their closest rival. In addition, considerable resources are spent determining structures and characterizing the activity of candidate structures as a pharmaceutical company hones in on a potential drug. This wealth of structure-activity data could be used to considerably improve the predictive power of the scoring function. However, since current ligand design packages employ their own proprietary scoring functions, this data is lost.
RACHEL offers the unique ability to utilize this structure-activity data and retain the competitive edge gained through research and development. By incorporating the necessary statistical and analytical tools, RACHEL allows the user to easily generate focused scoring functions to estimate ligand binding to a specific target receptor using proprietary structure-activity data. This allows companies who have characterized the receptor binding of a number of lead compound derivatives to utilize this knowledge in the design of future drugs.
The advantage of using focused scoring functions is significant. By limiting the training set to structures binding within the same receptor, we bias the scoring function towards the interactions that govern ligand association with the target active site. If hydrophobic contacts predominate, the hydrophobic descriptors will be emphasized. Conversely, if electrostatic forces are important to binding, those descriptors will be accentuated. Even something as simple as the size of the active site can have a tremendous impact on the allowable ligands. This is a descriptor that would be lost given a multitude of different training set receptors. As such, focused scoring functions have far more predictive power with respect to estimating ligand-receptor binding than generalized scoring functions.
Automated Target Function Elucidation
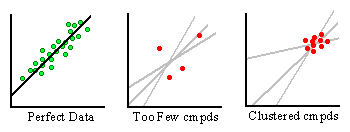
Figure 15B. Problems in deriving scoring functions.
Even with structure-activity data pertaining to a target receptor, difficulties in generating accurate scoring functions may arise as depicted in Figure 15B. First, there must be an adequate number of compounds to make the analysis statistically valid. Imagine the green and red dots to be structure-activity data points for individual ligand-receptor complexes. The lines passing through them represent potential scoring functions attempting to describe their distribution. In the graph on the left, we see an ideal distribution of complexes that allows for an easy determination of a best-fit line. This dataset contains a large number of complexes whose activity covers a wide range of values. A scoring function generated from this set thoroughly represents the data. The middle graph is more representative of the situation in academic research. Here we have far too few compounds to generate an accurate fit of the data. Notice the ambiguity that exists in determining the best-fit line. In essence, any scoring function derived from this dataset has little predictive value.
The graph on the right is another scenario that might occur. Here, there is no lack of data. However, given money and time constraints in drug development projects, it can be difficult to justify crystallographic studies on poorly binding compounds. As such, crystal structures of compounds are usually determined only when high affinity structures have been found by assay. Therefore, a cluster of high-affinity data points is produced. As one can see from the graph, it is also difficult to elucidate an accurate scoring function when the structure activity data is not broad enough.
In situations when the dataset is either too small or too clustered, RACHEL offers another means of generating a focused scoring system from proprietary structure activity data. If RACHEL determines that the derived scoring function offers little predictive value, she will revert to a target function. A target function is formed by simply averaging the descriptor values of the highest affinity training set complexes. These 'ideal' descriptor values are then used as a guide to determine if newly generated derivative structures are to be kept or discarded. This is illustrated in Figure 15C below.
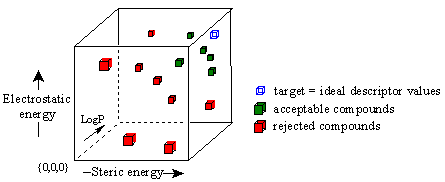
Figure 15C. Use of target function to screen compounds.
In this three-dimensional graph, the axes represent the three descriptors of ligand receptor binding we have referred to above. The blue cube is a plot of the 'ideal' descriptor values that have been averaged from the optimal binding ligands in the user's structure-activity data. This blue cube represents the target values against which all derivative compounds will be compared. The descriptor values for each derivative structure are then plotted. Those structures whose descriptor values are closest to the target, shown in green, are retained. All other structures are rejected.
The primary advantage of using a target function is its ease of implementation. No longer is a large training set of compounds required. Even a single compound can be used as a model for optimal ligand receptor binding. By simply extracting the descriptor values of the best compounds, we avoid many of the pitfalls in scoring function development that result from data artifacts. In addition, the characteristics of the ligand-receptor association that foster improved binding are allowed to drive the development of future structures.
The big disadvantage in using target functions is the lack of extrapolation. In other words, we are constraining the system using the properties of previously characterized ligands. Thus, we are unable to predict whether a new derivative compound can potentially bind better to the receptor than our best compounds. We are also unable to quantitate the binding relative to the other structures in the training set. We are simply building structures that mimic the characteristics of the best compounds.
Fortunately, this is often the exact task at hand for pharmaceutical chemists. By the time a drug development project has reached maturity, the ligands that have been developed are often optimal binding compounds. Therefore, a target function is usually sufficient as it allows the drug designer to construct alternate chemical architecture that retains optimal binding characteristics.