As discussed above, the development of any potential drug begins with years of scientific study to determine the biochemistry behind a medical problem for which pharmaceutical intervention is possible. The result is the determination of specific receptor targets that must be modulated to alter their activity in some way. Once these targets have been identified, the goal is then to find compounds that will interact with the receptors in some fashion. At this initial stage of drug development, it does not matter what effect the compounds have on the targets. We simply wish to find anything that binds to the receptor in any fashion.
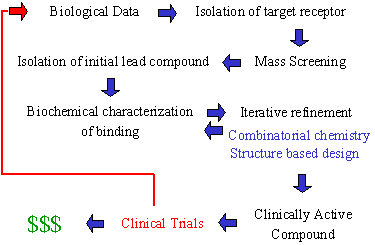
The modern day drug discovery pipeline is outlined in Figure 5. The first step is to determine an assay for the receptor. An assay is a chemical or biological test that turns positive when a suitable binding agent interacts with the receptor. Usually, this test is some form of colorimetric assay, in which an indicator turns a specific color when complementary ligands are present. This assay is then used in mass screening, which is a technique whereby hundreds of thousands of compounds can be tested in a matter of days to weeks. A pharmaceutical company will first screen their entire corporate database of known compounds. The reason is that if a successful match is found, the database compound is usually very well characterized. Furthermore, synthetic methods will be known for this compound, and patent protection is often present. This enables the company to rapidly prototype a candidate ligand whose chemistry is well known and within the intellectual property of the company.
Fig. 5. Drug discovery pipeline (circa 2001).
If a successful match is found, the initial hit is called a lead compound. The lead compound is usually a weakly binding ligand with minimal receptor activity. The binding of this structure to the receptor is then studied to determine the interactions that foster the ligand-receptor association. If the receptor is water soluble, there is a chance that x-ray crystallographic analysis can be employed to determine the three-dimensional structure of the ligand bound to the receptor at the atomic level. This is a very powerful tool for it allows scientists to directly visualize a snapshot of the individual atoms of the ligand as they reside within the receptor. This snapshot is referred to as a crystal structure of the ligand-receptor complex. Unfortunately, not all complexes can be analyzed in this manner. However, if a crystal structure can be determined, a strategy can then be developed based upon this characterization to improve and optimize the binding of the lead compound. From this point onward, a cycle of iterative chemical refinement and testing continues until a drug is developed that undergoes clinical trials. The techniques most often used to refine drugs are combinatorial chemistry and structure based design.
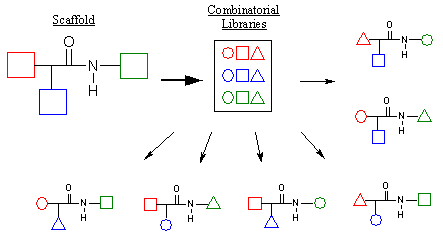
Figure 6. Combinatorial chemistry schematic.
Combinatorial chemistry is a very powerful technique that chemists can employ to aid in the refinement of the lead compound. Combinatorial chemistry is a synthetic tool that enables chemists to rapidly generate thousands of lead compound derivatives for testing. As shown above in Figure 6, a scaffold is employed that contains a portion of the ligand that remains constant. Subsite groups (shown in red, green, and blue) are potential sites for derivatization. These subsites are then reacted with combinatorial libraries to generate a multitude of derivative structures, each with different substituent groups. One can see how a vast number of compounds can be generated as a result of the combinatorial process. If a scaffold contains three derivatization sites and the library contains ten groups per site, theoretically 1000 different combinations are possible. By carefully selecting libraries based upon the study of the active site, we can target the derivatization process towards optimizing ligand receptor interaction.
![]()
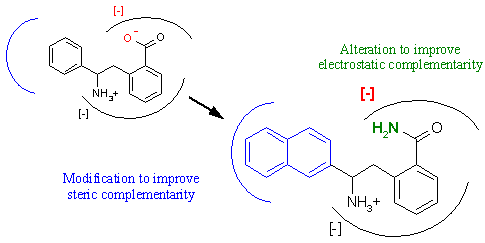
Structure based design, often called rational drug design, is much more focused than combinatorial chemistry. As shown above in Figure 7, it involves using the biochemical laws of ligand-receptor association discussed above to postulate ligand refinements to improve binding. For example, we discussed that steric complementarity is vital to tight receptor binding. Using the crystal structure of the complex, we can target regions of the ligand that fit poorly within the active site and postulate chemical changes to improve complementarity with the receptor. In a similar fashion, functional groups on the ligand can be changed in order to augment electrostatic complementarity with the receptor. However, the danger in altering any portion of the ligand is the effect on the remaining ligand structures. Modifying even a single atom in the middle of the ligand can drastically change the shape of the overall structure. Even though complementarity in one portion of the ligand might be improved by the chemical revision, the overall binding might be severely compromised. This is the difficulty in any ligand refinement procedure.
Prev - The Challenge of Drug
Design
Next - Use of Computers in Drug Design
Return to RACHEL Technology - Main
(c) 2002 Drug Design Methodologies, LLC. All Rights Reserved