Since the 1990s, dozens of rational drug design packages have been published. These programs fall in one of three main genres: 1. Scanners, 2. Builders, or 3. Hybrids.
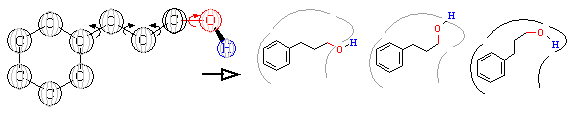
Figure 10A. Ligand flexibility.
Ligand flexibility is a concern in
builder-type programs. The ligand analogy to a key discussed above is slightly
misleading. While a key is a static unbending object, a chemical structure
is constantly changing shape. As shown in Figure 10A, a molecule is
actually composed of rigid chemical groups separated by rotatable bonds, as
defined by the laws of chemistry. These rotatable bonds give ligands
inherent flexibility. In the right side of this figure, we see how a
ligand can adopt numerous configurations as it attempts to bind within the
active site. Given a newly generated compound, the big challenge is to
computationally search all the potential configurations to determine if binding
is possible. If so, the task is then to determine the most complementary
binding structure.
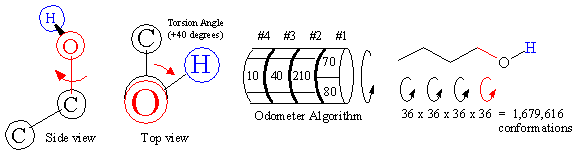
Figure 10B. Combinatorial nature of conformational searching.
Although accurately calculating ligand receptor affinity remains the most scientifically challenging endeavor in drug design, searching all the rotatable bond configurations a ligand can adopt is the most demanding. A snapshot structure of the ligand at any instant in time is called a conformation, and is defined by the set of torsion angles between rigid groups. As shown in Figure 10B above, each rotatable bond can potentially spin 360 degrees, and the two attached groups define the torsion angle. The difficulty in determining all possible conformations lies in the combinatorial nature of the problem. Because each bond can potentially sweep the entire arc, an 'odometer' algorithm must be employed.
The odometer algorithm is a systematic
sampling of all possible torsion angle combinations. Like an odometer, the
first bond is fully rotated 360 degrees before the second bond is
incremented. When the second bond is incremented, the first bond is reset
and then fully scanned again. This continues until the second bond is fully
rotated, at which time the third bond is incremented. Searching continues
in this manner until all rotatable bond combinations are eventually sampled as
shown above.
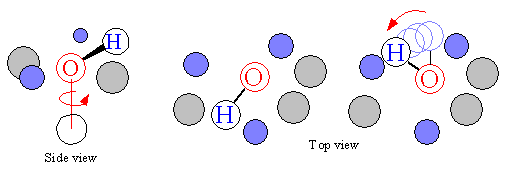
Figure 10C. Determining valid conformer torsion angles.
During the conformational search, acceptable torsion angle ranges must be elucidated for each rotatable bond. This is shown in Figure 10C. When a rotatable bond is incremented, the atoms attached to the 'swing arm' are checked against all receptor atoms and ligand groups within the vicinity. If contact exists, then that particular conformer is eliminated. As such, only valid ligand conformations that conform to the active site are generated.
As shown in Figure 10B above, the combinatorial nature of this problem leads to an exponential rise in the number of conformations that must be calculated. A four rotatable bond ligand sampled at ten-degree increments adds up to 1,679,616 different conformations. However, a five rotatable bond ligand sampled at ten-degree increments includes over 60 million conformations! Considering that most drugs contain 10 - 15 rotatable bonds, this value can easily overwhelm even the fastest computers.
Algorithms have been developed that reduce the computational burden of conformational searching by orders of magnitude. Nevertheless, commercial builder packages implement conformational flexibility in different ways to compensate for the associated computational burden. Some do not implement conformational flexibility at all, which severely limits their ability to determine adequate ligand binding conformations. Others use rudimentary, pre-calculated torsion angle scans that lack the resolution to tightly dock compounds within the active site. Only packages that implement a systematic conformational search are able to sufficiently determine potential binding conformations.
Prev - Builders
Next - Hybrids
Return to RACHEL Technology - Main
(c) 2002 Drug Design Methodologies, LLC. All Rights Reserved