RACHEL stands for Real-time Automated Combinatorial Heuristic Enhancement of Lead compounds and is a drug optimization package written by Chris M.W. Ho, M.D.Ph.D. of Drug Design Methodologies, LLC. This program is designed to optimize weak binding lead compounds in an automated, combinatorial fashion. From our discussions above, this software would be classified as a 'builder-type' drug refinement program. With our brief introduction into the basics of drug design, we can now discuss the technological advances that make RACHEL superior to all current computer-aided drug refinement software packages.
We have discussed how builder-type programs work. In short, a database of chemical fragments is used to derivatize a lead compound by replacing weak binding regions with components that will improve receptor complementarity. These compounds are then scored by calculating their affinity for the receptor. Those compounds that bind tightly with the receptor are then saved while those that bind poorly are discarded. This new population of compounds is then processed to form the next generation of derivatives. Over time, a lead compound is iteratively refined into a set of tight binding structures.
When using current commercial ligand refinement packages of this type, the user is dependent on the software for several vital functions that are critical for drug development success. First and foremost, a database of chemical building components is required. All current commercial packages provide this database of components, which allows any researcher to immediately use these tools for drug design. However, this is not always desirable. Pharmaceutical firms are always competing against rival companies. Intellectual property in the form of patented database structures, synthetic know-how, and the biochemical data of characterized lead compounds provides a competitive edge against other companies. As such, using a program that contains both a standard database and a standard scoring function offers no such advantage over another company.
RACHEL avoids this shortcoming by allowing the company (or academic laboratory) to utilize its intellectual property in the design of new drugs. RACHEL pulls building block components directly from the user’s corporate database. As such, companies who have invested considerable time and money developing the chemistry to synthesize a particular class of compounds can utilize this knowledge in the design of future drugs. This has the added benefit that generated structures are easier to patent as proprietary chemistry is incorporated. This is a tremendous advantage to using our software, and no other design package employs this feature.
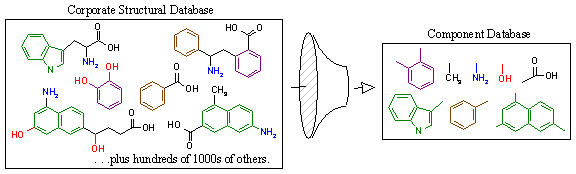
Figure 12A. Extraction of components from corporate structural database.
Figure 12A schematically demonstrates this process. On the left, we have the corporate structural database, which may contain hundreds of thousands of compounds. All structures are composed of non-rotatable chemical groups separated by rotatable bonds as defined by the laws of chemistry. These non-rotatable groups represent the fundamental building blocks that will be used to regenerate new derivative compounds. RACHEL first isolates these components by identifying the rotatable bonds in the structure as shown below in Figure 12B (red arrows). The individual components are then isolated, identified with a unique label that describes its distinct chemical architecture, and stored in the component database along with a description of its chemical composition. The component label is very important as it is used to register each fragment and prevent the storage of redundant chemical groups.
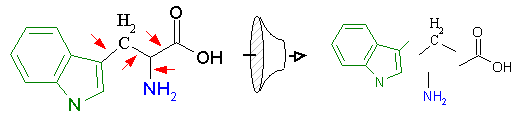
Figure 12B. Separation and isolation of components at rotatable bonds.
There are numerous advantages to extracting components in the manner described above. First, the storage of unique components allows the compression of a massive corporate database into a much smaller and manageable form. Typically a corporate database containing 100,000 structures may be comprised of only 5,000 individual components. This is because a few select components, such as methyl, hydroxyl, and amine groups, are utilized over and over again. Second, unique chemical constructions, for which only proprietary synthetic methods are known, are stored and available for use in future ligand design. This allows the user to take advantage of patented corporate chemistry and preserve the competitive edge gained from prior research.
The goal of builder-type programs is to generate derivatives that are complementary to the active site. Both steric (size and shape) as well as electrostatic forces must be considered. The difficulty in accomplishing this lies in the sheer number of potential component combinations that are possible. As a result, nearly all commercial packages randomly select fragments for assembly. While this ensures an adequate sampling of components, it often leads to the selection of improper fragments. As such, many iterations of structure refinement are wasted generating poor derivatives.
The RACHEL software has a far greater problem. While current builder-type software packages contain databases with 100 components or less, RACHEL can extract upwards of 40-50,000 components depending upon the size and diversity of the corporate database. Thus, the number of potential fragment combinations is nearly immeasurable. Clearly, a method is needed to rapidly focus on the appropriate combinations that are likely to satisfy binding requirements.
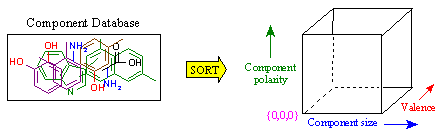
Figure 13A. Generation of Diversity Index.
The greatest benefit of RACHEL's component extraction method is that a massive diversity index of the entire corporate database is created. Along with the atomic coordinates of each component, a wealth of chemical information characterizing each building block is stored. Data such as the size of the component, atom composition, connectivity, ring structure, and electrostatic charge are included. As such, a means of rapidly cross-referencing chemical components on demand is available.
Figure 13A demonstrates how this diversity index is generated. On the left, we see a representative component database. Using the stored chemical attributes, the database is sorted and mapped into a multi-dimensional array, where each axis represents a different descriptor. In this example, we only show size, polarity, and valence (number of connections) for simplicity. Each axis provides a gradient along which components can be distinguished. As such, components that are similar with respect to the various descriptors are grouped together.
This diversity index offers a powerful means to improve the generation of complementary ligands. Over time, builder-type programs evolve compounds with improved binding. A moderate affinity structure has reasonable steric and electrostatic complementarity with the active site. However, components can still be added, deleted, or substituted to augment receptor interaction.
As stated above, nearly all commercial builder-type programs select substituent fragments at random. Although simplistic, this is absolutely necessary to ensure adequate sampling of the database and generate truly novel solutions. RACHEL implements random sampling in the initial stages of lead compound optimization. Early derivatives that are generated are weak binding at best. Thus, random component sampling increases the chances of finding the appropriate components to improve receptor interaction.
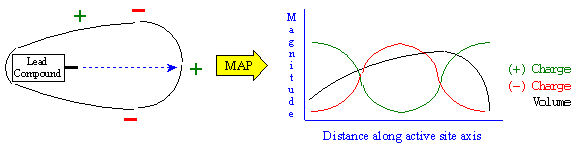
Figure 13B. RACHEL Active Site Mapping.
However, random sampling often diminishes the complementarity of reasonable binding compounds. This is the result of replacing satisfactory components with poor ones. For example, if a small methyl group or a highly charged fragment were to replace a large, hydrophobic ring on the ligand, it would ruin interaction with the receptor at that component. Instead, RACHEL incorporates a heuristic active site mapping algorithm as shown in Figure 13B to determine the optimal chemical characteristics to complement a given region of the active site. This technique maps chemical characteristics of the receptor, such as positive charge, negative charge, and active site volume as a function of distance along the active site axis. Using this active site map, RACHEL can determine the chemical characteristics most likely to complement the receptor at a given component location. RACHEL then determines a list of candidate fragments and substitutes them in a combinatorial fashion.
This is diagrammed in Figure 13C below. In this example, RACHEL determines that the naphthalene group (blue) and carboxylic acid group (red) of a ligand derivative should be replaced with other components to improve binding. The naphthalene group is large and very non-polar since it is strictly hydrocarbon. Conversely, the carboxylic acid group is quite small, but highly polar. Using the active site map as described above, RACHEL determines that these characteristics are indeed ideal for complementing the receptor at each respective component. Using the diversity index, RACHEL can cross-reference other database components that exhibit similar characteristics, as shown in the red and blue boxes on the right. These components are then combinatorially used to generate a new family of derivatives for testing. Each derivative retains the optimal receptor binding characteristics. However, enough variability is generated to potentially improve receptor complementarity.
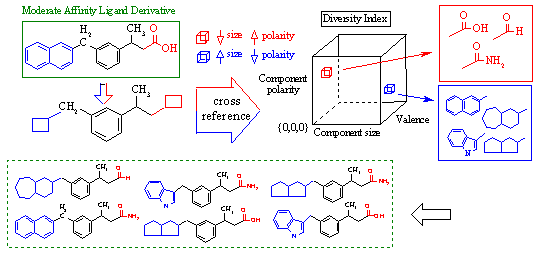
Figure 13C. RACHEL Intelligent Component
Selection System.
Prev - Hybrids
Next - Component Language
Return to RACHEL Technology - Main
(c) 2002 Drug Design Methodologies, LLC. All Rights Reserved