As we discussed above, the calculation of receptor binding affinity for each newly generated derivative ligand remains the most challenging aspect of drug design. Not only is this task very difficult, it also is critical for the success of the program. If the calculated binding affinities have no bearing on reality, then, the program might as well generate random structures. The big difficulty is that the accurate determination of ligand receptor binding involves complex, intensive, quantum chemical calculations, which can take days to weeks on even the fastest computers. On the other hand, a typical ligand refinement program can generate and sample new structures at the rate of several hundred to thousands a minute. Thus, in order to achieve such high throughput, there must be some compromise in the accuracy of the binding calculation. That compromise is in the utilization of scoring functions.
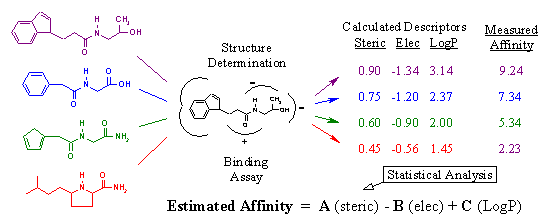
Figure 15A. Derivation of scoring function.
Scoring functions estimate ligand-binding affinity using descriptors that can be rapidly measured from the ligand receptor interaction. In essence, a scoring function is an equation that relates measurable descriptors of binding to ligand receptor affinity. Figure 15A reveals how scoring functions are derived. Given a particular ligand and receptor, two things must be done. First, the ligand must be reacted with the receptor to determine its actual binding affinity using a biological assay. Second, the three-dimensional structure of the ligand bound within the receptor must be determined using x-ray crystallography. As we discussed above, the determinants of binding include steric interaction energy, electrostatic interaction energy, and hydrophobicity. Given the three-dimensional structure of a particular compound bound within the active site, we can rapidly calculate the values for these descriptors. For review - see Head, R.D. et. al, J. Am. Chem. Soc., 118: 3959-3969 (1996).
For example, to calculate steric interaction energy, we simply sum up the number of receptor atoms that are within a specific distance (i.e. 5 angstroms) away from any ligand atom. The higher the value, the more interaction between ligand and receptor atoms. Electrostatic interaction energy is computed using Coulomb’s law, which can be found in any high school physic book. Hydrophobicity is represented by LogP, which is a measure of the compound’s propensity to solubilize in oil versus water. The higher the value, the more greasy and oily the compound is. In short, these descriptors are simple and very easy to calculate. This allows for the rapid determination of characteristics that relate to ligand binding strength.
In our figure, we have four complexes whose binding affinity has been measured and whose descriptors have been calculated. Statistical tools, such as partial least squares regression, are then employed to generate the equation relating the numerical trends in the descriptors with the corresponding binding affinities. What results is an equation where estimated affinity is a function of the calculated descriptors (steric, electrostatic, and logP) multiplied by their corresponding coefficients (A, B, and C). It is the coefficients that relate the calculated descriptors to the actual affinities, and are determined by the statistical analysis. For example, we can see that as steric interaction energy increases, so does the biological binding activity. Thus, the coefficient (A) is positive. On the other hand, we know that a negative electrostatic interaction energy is conducive to tighter binding since opposite charges attract. Therefore, the corresponding coefficient (B) is negative. LogP follows a similar trend as steric interaction energy; thus, coefficient (C) is positive.
Once a scoring function has been derived, it can be employed to estimate binding affinities very rapidly. Given a newly designed ligand or structural derivative that has been docked within the active site, the descriptors of binding are first calculated. These descriptors are then multiplied by the derived coefficients of the scoring function. Once all the terms have been calculated, they are then summed to determine the estimated binding affinity of the ligand in question. It is important to note that this example is very simplistic. In reality, some scoring functions contain over twenty terms. Table 7 lists the ligand receptor binding descriptors employed by RACHEL.
|
Steric complementarity. |
Molecular weight. |
|
Steric strain. |
Number of rotatable bonds. |
|
Electrostatic interaction energy. |
LogP estimation. |
|
Nonpolar - nonpolar interaction energy. |
Nonpolar atom fraction. |
Table 7. RACHEL descriptors used to generate scoring functions.
Currently, there are several hundred high quality ligand-receptor complexes in the public domain that can be employed for scoring function development. Pharmaceutical firms have access to far more proprietary structures. However, even with all these structures along with the powerful statistical tools to analyze them, scoring functions still remain mediocre at best. According to the laws of thermodynamics, DG = DH - TDS. DG is the Gibbs free energy of binding, and is the energy that is released when ligand and receptor bind. This is the actual thermodynamic property that we are trying to estimate with the scoring function. DH is enthalpy (internal energy), and is grossly approximated by the calculated descriptors. Efforts to improve the accuracy of these approximations often increase calculation time drastically. TDS is an entropy term, and is indicative of the relative gain or loss of disorder when ligand and receptor bind. Perhaps the biggest influence on entropy is the behavior of the water molecules in the active site that are displaced when binding occurs. This is often disregarded, as it is difficult to accurately calculate without considerable computation. Thus, the take-home message is that DG is at best very crudely estimated by any scoring function. For review - see Ajay and Murcko, J. Med. Chem., 38: 4953-4967 (1995).
Compounding this problem is the fact that all current commercial packages utilize a single, proprietary, generalized scoring function that has been derived using a wide variety of structures. There are two significant problems with this approach. First, receptor systems vary considerably in their chemical makeup. In some systems, electrostatic interactions dominate the ligand binding force. In other systems, hydrophobic interactions overshadow the other forces involved. Thus, a master scoring function to estimate binding affinity for all ligand receptor systems becomes a 'jack of all trades, master of none'. Using such a variety of ligand receptor systems in the training set adds considerable noise to the data, which diminishes its predictive power.
The second shortcoming in using a generalized scoring function is the loss of competitive advantage. In effect, any laboratory or company that employs these tools utilizes the same predictive function as their closest rival. In addition, considerable resources are spent determining structures and characterizing the activity of candidate structures as a pharmaceutical company hones in on a potential drug. This wealth of structure-activity data could be used to considerably improve the predictive power of the scoring function. However, since current ligand design packages employ their own proprietary scoring functions, this data is lost.
RACHEL offers the unique ability to utilize this structure-activity data and retain the competitive edge gained through research and development. By incorporating the necessary statistical and analytical tools, RACHEL allows the user to easily generate focused scoring functions to estimate ligand binding to a specific target receptor using proprietary structure-activity data. This allows companies who have characterized the receptor binding of a number of lead compound derivatives to utilize this knowledge in the design of future drugs.
The advantage of using focused scoring functions is significant. By limiting the training set to structures binding within the same receptor, we bias the scoring function towards the interactions that govern ligand association with the target active site. If hydrophobic contacts predominate, the hydrophobic descriptors will be emphasized. Conversely, if electrostatic forces are important to binding, those descriptors will be accentuated. Even something as simple as the size of the active site can have a tremendous impact on the allowable ligands. This is a descriptor that would be lost given a multitude of different training set receptors. As such, focused scoring functions have far more predictive power with respect to estimating ligand-receptor binding than generalized scoring functions.
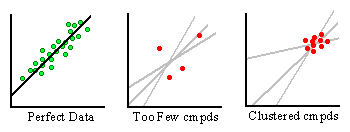
Figure 15B. Problems in deriving scoring functions.
Even with structure-activity data pertaining to a target receptor, difficulties in generating accurate scoring functions may arise as depicted in Figure 15B. First, there must be an adequate number of compounds to make the analysis statistically valid. Imagine the green and red dots to be structure-activity data points for individual ligand-receptor complexes. The lines passing through them represent potential scoring functions attempting to describe their distribution. In the graph on the left, we see an ideal distribution of complexes that allows for an easy determination of a best-fit line. This dataset contains a large number of complexes whose activity covers a wide range of values. A scoring function generated from this set thoroughly represents the data. The middle graph is more representative of the situation in academic research. Here we have far too few compounds to generate an accurate fit of the data. Notice the ambiguity that exists in determining the best-fit line. In essence, any scoring function derived from this dataset has little predictive value.
The graph on the right is another scenario that might occur. Here, there is no lack of data. However, given money and time constraints in drug development projects, it can be difficult to justify crystallographic studies on poorly binding compounds. As such, crystal structures of compounds are usually determined only when high affinity structures have been found by assay. Therefore, a cluster of high-affinity data points is produced. As one can see from the graph, it is also difficult to elucidate an accurate scoring function when the structure activity data is not broad enough.
In situations when the dataset is either too small or too clustered, RACHEL offers another means of generating a focused scoring system from proprietary structure activity data. If RACHEL determines that the derived scoring function offers little predictive value, she will revert to a target function. A target function is formed by simply averaging the descriptor values of the highest affinity training set complexes. These 'ideal' descriptor values are then used as a guide to determine if newly generated derivative structures are to be kept or discarded. This is illustrated in Figure 15C below.
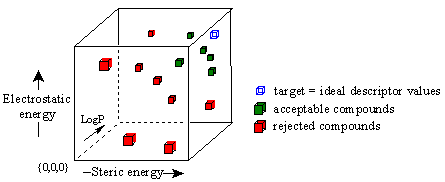
Figure 15C. Use of target function to screen compounds.
In this three-dimensional graph, the axes represent the three descriptors of ligand receptor binding we have referred to above. The blue cube is a plot of the 'ideal' descriptor values that have been averaged from the optimal binding ligands in the user's structure-activity data. This blue cube represents the target values against which all derivative compounds will be compared. The descriptor values for each derivative structure are then plotted. Those structures whose descriptor values are closest to the target, shown in green, are retained. All other structures are rejected.
The primary advantage of using a target function is its ease of implementation. No longer is a large training set of compounds required. Even a single compound can be used as a model for optimal ligand receptor binding. By simply extracting the descriptor values of the best compounds, we avoid many of the pitfalls in scoring function development that result from data artifacts. In addition, the characteristics of the ligand-receptor association that foster improved binding are allowed to drive the development of future structures.
The big disadvantage in using target functions is the lack of extrapolation. In other words, we are constraining the system using the properties of previously characterized ligands. Thus, we are unable to predict whether a new derivative compound can potentially bind better to the receptor than our best compounds. We are also unable to quantitate the binding relative to the other structures in the training set. We are simply building structures that mimic the characteristics of the best compounds.
Fortunately, this is often the exact task at hand for pharmaceutical chemists. By the time a drug development project has reached maturity, the ligands that have been developed are often optimal binding compounds. Therefore, a target function is usually sufficient as it allows the drug designer to construct alternate chemical architecture that retains optimal binding characteristics.
Prev - Component Specification
Language
Return to RACHEL Technology - Main
(c) 2002 Drug Design Methodologies, LLC. All Rights Reserved